
Overview
The past twenty-five years has seen a rapid decrease in the cost of genetic sequencing, from $2.7 billion dollars for the human genome project (completed 2003), to roughly $1000 dollars today. This decrease in cost has led to the development of the personal genomics testing via companies like 23andMe and AncestryDNA, who provide these testing services directly to the public, albeit, using a cheaper variant testing method rather than whole genome sequencing.
Historically, the volume of information provided by genetic testing presented a hurdle for any analyst, as only universities, private research institutes or computer science labs had the necessary equipment to process the large datasets in reasonable amounts of time. The advent of cloud computing, and in particular cloud-scale data warehouses such as Google BigQuery or Redshift, now puts this capability in the hands of small startups and individual users at home.
I recently obtained a genetic test which included access to the raw Single Nucleotide Polymorphism (SNP) data. The test itself was for heritage, however armed with the data and a GCP account, I wanted to see what I could glean from the results.
Note:
For anyone actually considering taking a test such as this, make sure you consider the potential ramifications, including discovering information about yourself for which you may not be prepared. It’s also worth considering the impact of any tests on disclosure requirements for health and life insurance. Additionally, this is one area where it’s really important to read the terms and conditions concerning how the testing parties will handle your sample and data, how they’ll store and/or use the data.
Most genetic lineage tests (including the one I obtained) will provide a raw dump of the data, however this is often not prepared and tested to the same standard as a medical genetic test, and accordingly should not be acted upon without additional confirmation.
Brief Biology Explanation
For those of you who didn’t take high school biology (or are a bit rusty!), I’ve provided a little bit of a primer to get you familiar with the underlying science.
Your genome is the genetic material that makes you, you, minus all the environmental and social effects. It includes not only your DNA, the twisted helix you see everywhere, but also the genetic material of mitochondria (of Star Wars fame) and chloroplasts. For the sake of brevity and this exercise, we’ll focus on only the DNA part, though most heritage tests will provide mitochondria tests for matrilineal ancestry.
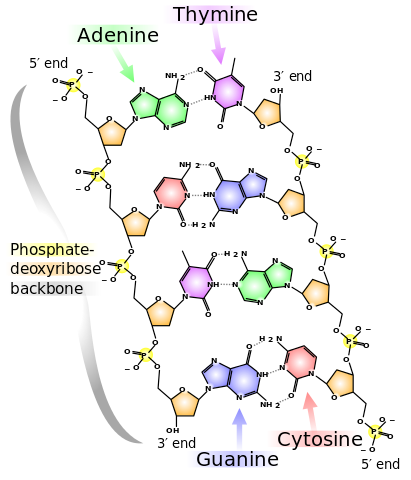
DeoxyriboNucleic Acid (DNA, shown to the right) comprises two strands called polynucleotides, polynucleotide is just a fancy way of saying many-nucleotides. A nucleotide comprises of one molecule called a nucleobase (base for short) which can be any one of adenine (A), cytosine (C), guanine (G) or thymine (T) as well as a sugar and phosphate group. In DNA the bases pair selectively, A will only bind with T and C will only bind with G. A pair of bound bases is called a base-pair (bp).
The selective pairing of bases means that a sequence on one strand, such as ACTG, will have a complementary pair on the other strand: TGAC. These pairs of nucleobases (with the sugar and phosphate groups) are what makes up DNA, and DNA makes up your chromosomes, which are tightly-packed bundles of DNA that exists within your cells. Human cells ordinarily contain 23 pairs of chromosomes (22 called autosomes and a pair of sex chromosomes – XX or XY).
Returning to the situation at hand, a single nucleotide polymorphism (greek for ‘many’-‘shape’ roughly) is when a single base-pair exhibits multiple variations at a particular site in the DNA strands – think A-T instead of G-C. This can be benign, pathogenic, beneficial or unknown. Each variation is called an allele, typically a particular allele is the more ‘normal’ common pairing and the other variations are rarer. It’s worth also noting that A-T is not the same as T-A, as the strands have direction and are anti-parallel (parallel but heading in different directions).
A genetic test such as those provided by the commercial testing companies will detect and identify these SNPs and use them to provide an indication of potential medical impacts and/or to trace lineage (as you inherit your DNA from your mother and father).
Process
The company I used for testing provided text file dumps of my autosomal sequence (22 chromosome pairs minus the sex chromosomes), my Y chromosome (as I am male I have an X and Y chromosome, females will have XX) and a sequence of my mitochondrial DNA (this is inherited matrilinealy and can trace your motherline). Only the autosomal sequence provided the SNP detail, the other two files were lists of haplotypes which are more tailored for lineage analysis.
The first step was to get the data into a suitable format for ingestion. Most tools have centered around the Variant Call Format (VCF) for representing data, so first step was to use a tool called ‘PLINK’ to convert from the text file format to VCF. I uploaded the resulting VCF into a Cloud Storage bucket under a Genomics project I had created for this work.
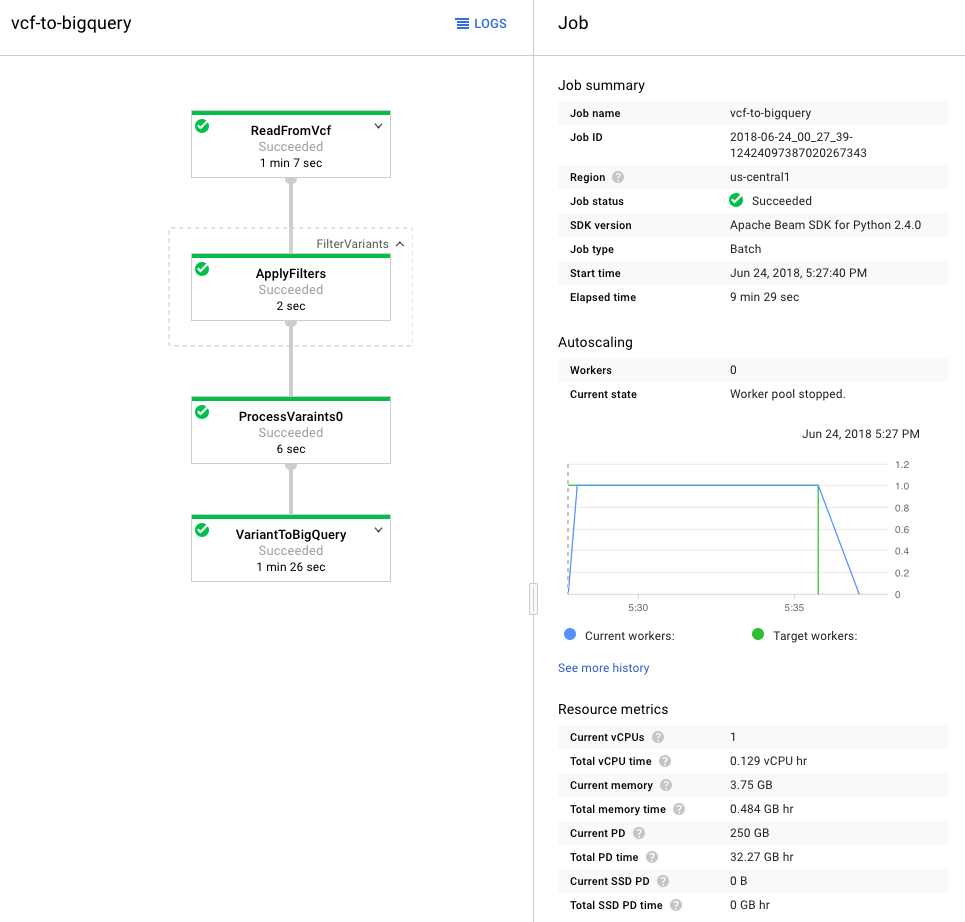
Once processed, I used the GCP Variant Transforms tool to transform and process the VCF file. This tool uses the Google Genomics Pipelines API to run a pre-built docker image using Google Dataflow. The docker image contains all the binaries and dependencies required for reading, filtering, processing and loading the VCF files into BigQuery. This step took around 10 minutes to load a 25MB file into BigQuery (shown below). The resulting table was just over 600k rows and 38MB.
In order to annotate my data, I needed a dataset that I could compare my SNPs against. Fortunately, Tute Genomics has made available a human reference genome complete with annotations, including clinically significant SNPs. This dataset contains 9 billion records and weighs in at 649GB so manipulating it on my MacBook Pro would have increased its internal temperature somewhat.
My goal was to run a table join that would match my SNPs with those in the Tute Genomics dataset and output the results to a separate (smaller) table. I’d then be able to peruse the results for interesting findings. I was able to modify an example query from Google’s bigquery-examples to join my data against Tute Genomics. This processed 328GB of data in 10.2 seconds.
Findings
I found 243 SNPs of various medical significance. I already had a fair understanding of my family medical history so a lot of the findings confirmed what I suspected. For example, it did confirm I’m susceptible to melanomas, which I knew from family history (relatives have had them removed) and I get checks on a regular basis. I did find that I have gene that gives an increased response to opoids, so I should avoid heroin, which is generally good advice, genes or not.
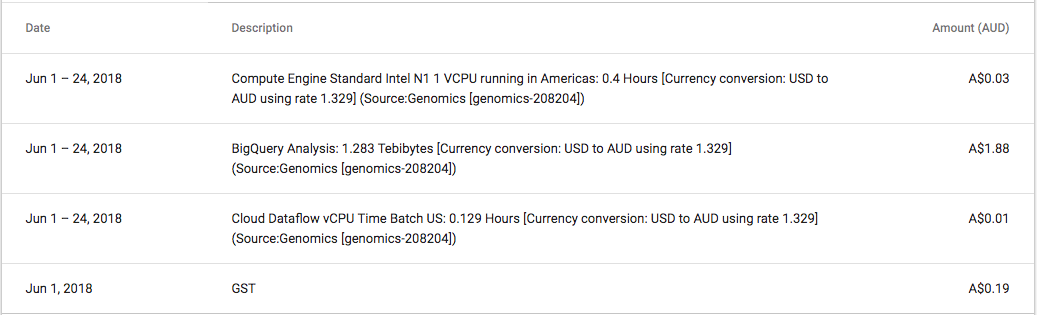
One thing that did ‘wow’ me was the speed of BigQuery, and the huge datasets publicly available. Being able to process huge volumes of data and tweak queries whilst receiving almost immediate feedback is amazing. The cost of this analysis ran up to $2, and that was because I ran the expensive query a couple of times to tweak the results.
The wealth of knowledge and guidance on the platform is also exceptional. I’m currently working through the Data Engineer course, so the amount of guides to rely upon was massively helpful. I’m by no means a SQL jockey, so the BigQuery interface was a godsend in being able to repeatedly tweak my queries.
It’s also worth noting that the APIs are still in alpha, and are documented as so, I tried using a couple of examples only to find that the API had been deprecated. Also, unlike Amazon, you have to ‘enable’ a lot of APIs before you can use them on GCP, so if you get ahead of yourself you can attempt to use a service before it’s enabled, which can be frustrating.
Improvements & scope for further work
This was very much a console-driven exercise, a proper implementation would definitely use automation to ingest the files from the Storage Bucket through to BigQuery, per Color’s blogpost.
Given the sensitivity of the data, it would also be interesting to investigate what controls we could put in place between datasets and how we would manage this data as either an organisation providing results to individuals, or an organisation providing data-enrichment ‘as a service’.
Summary
Overall this was a great way to work with established and developing Google services as well as have some fun in an area in which I have an interest. I look forward to further developments in this space and will definitely keep an eye on the Google Genomics project space.
References and further reading
Mike Kahn’s interpretion of 23andMe results: This is a great resource and got me started, just note that the APIs have been deprecated since he wrote this.
Google Genomics GCP Variant Transforms repository: GitHub repository for the Variant Transforms tool I used to load VCF data.
Google Genomics BigQuery Examples repository: GitHub repository containing many interesting BigQuery examples that I modified for this post.
Google Slide deck ‘Variant Transforms and BigQuery’: Google Cloud Healthcare and Life Sciences slide deck providing an overview of BigQuery and Dataflow pipelines.
PLINK tool: Tool I used to convert raw data to VCF format.

Brent Harrison is a Senior Consultant in Sourced Group's Sydney consulting practice. Joining the group in 2014 after leaving the science industry, Brent enjoys helping customers solve complex problems in novel and inventive manners.


